









Cos’è la granulomatosi eosinofila con poliangite (EGPA)?
La granulomatosi eosinofila con poliangite (EGPA), precedentemente nota come sindrome di Churg-Strauss, è una rara forma di vasculite ANCA-associata, caratterizzata da un'infiammazione granulomatosa ricca di eosinofili e da una vasculite che colpisce vasi di piccole e medie dimensioni, associata ad asma bronchiale ed eosinofilia.1 La prevalenza mondiale di EGPA è di 10,7-17,8 ogni milione di abitanti con variazioni in base all'area geografica e ai criteri considerati.1 La patogenesi di EGPA rimane in gran parte sconosciuta; tuttavia, come per molte malattie autoimmuni, si ritiene che fattori genetici e ambientali contribuiscano al suo sviluppo.1
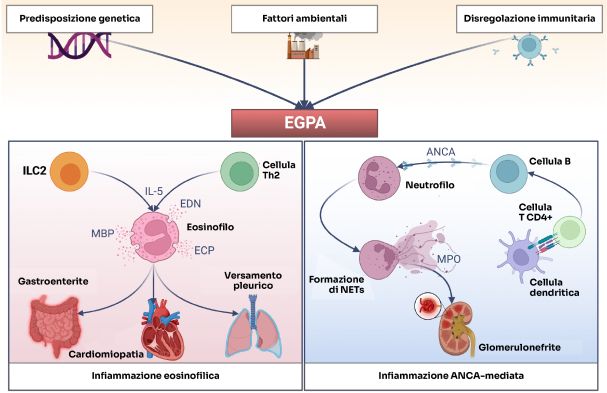
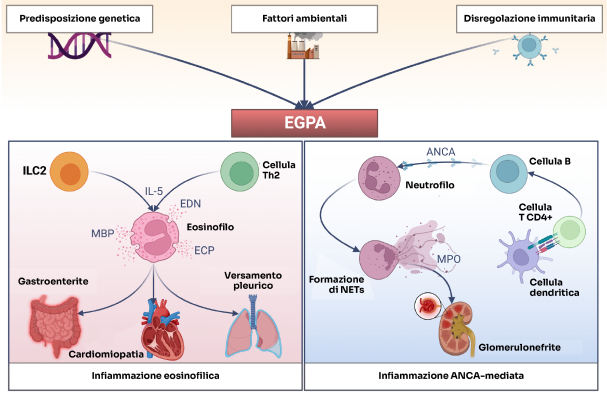
ANCA: anticorpi anti-citoplasma dei neutrofili; ECP: proteina cationica degli eosinofili; EDN: neurotossina derivata dagli eosinofili; ILC2: cellula linfoide innata di tipo 2; IL-5: interleuchina-5; MBP: proteina basica maggiore; MPO: mieloperossidasi; NETs: trappole extracellulari dei neutrofili; Th2: linfociti T helper di tipo 2.
Rielaborazione grafica da Fig. 2, Rif. 2
Caratteristiche cliniche della granulomatosi eosinofila con poliangite3
L'EGPA è considerata una malattia sistemica e tipicamente evolve in fasi prodromiche che possono parzialmente sovrapporsi e non seguire un ordine definito.3
- Fase allergica: caratterizzata da asma e rinosinusite cronica.3
- Fase eosinofilica: caratterizzata da eosinofilia periferica e coinvolgimento di diversi organi.3
- Fase vasculitica: caratterizzata da manifestazioni cliniche dovute a vasculite dei piccoli vasi.3
Il tasso di mortalità globale nei pazienti trattati con EGPA è pari al 20-25% nell’arco di 5-10 anni.4
Fattori prognostici sfavorevoli per la granulomatosi eosinofila con poliangite4
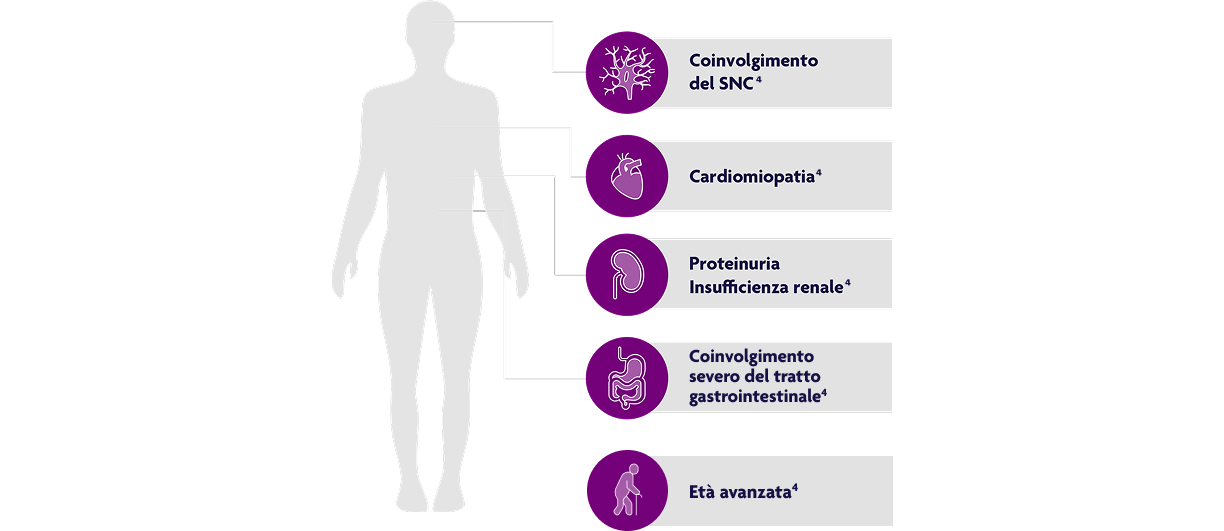
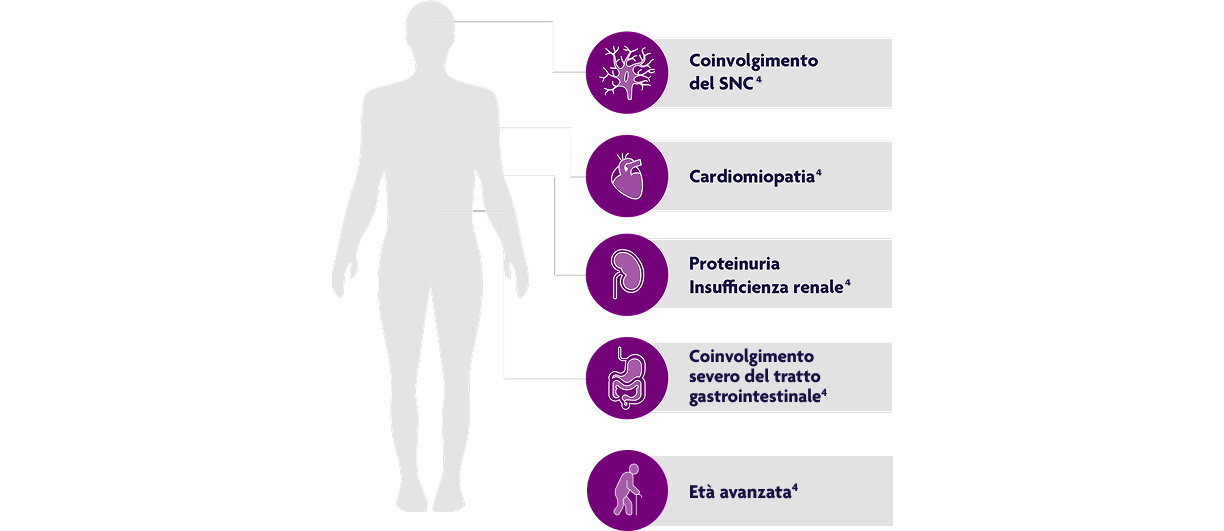
Elaborazione grafica da testo, Rif. 4.
EGPA: granulomatosi eosinofila con poliangite; SNC: sistema nervoso centrale.
Bibliografia
- Furuta S, et al. Allergol Int. 2019; 68 (4): 430-436.
- Carrón-Herrero A, et al. Explor Asthma Allergy 2023; 1: 31–48.
- Izquierdo-Domínguez A, et al. Sinusitis 2016, 1, 24-43.
- Taniguchi M, et al. Allergol Int 2007; 56 (2): 97-103.
Il portale di AstraZeneca nasce con l’intento di proporre uno spazio di dialogo digitale tra i medici, gli specialisti e l’azienda.
Un progetto:
